Фосфор (хим.; Phosphore франц., Phosphor нем., Phosphorus англ. и лат., откуда обозначение P, иногда — Ph; атомный вес 31 [В новейшее время атомный вес Ф. найден (van der Plaats) такой: 30,93 — путем восстановления определенным весом Ф. металлического серебра из соляного раствора; 31,01 — на основании анализа Ag3PO4, 30,98 — при взвешивании P2O5, полученного сжиганием определенного количества обыкновенного Ф. Последний метод, для красного Ф., дал в среднем из 10 опытов 31,025 (Schrötter, 1853). К этим данным близки результаты Берцелиуса 31,17—31,33, полученные при осаждении фосфором серебра и золота из соляных растворов.], если О = 16), как и сера, принадлежит к числу важнейших неметаллических элементов и по своим запасам на нашей планете даже превосходит ее — согласно расчетам Кларке, он составляет 0,09 %, а сера 0,04 % земной коры. В отличие от серы Ф. по великой своей окисляемости не встречается в природе в свободном виде, а исключительно в виде солей фосфорной кислоты, каковы фосфаты кальция, алюминия и железа — см. Фосфаты, Фосфориты. Он входит в состав животных и растительных тканей — см. Фосфор (аллотропия). В свободном виде Ф. впервые был получен алхимиком Брандом в Гамбурге из мочи в 1669 г., для чего он выпаривал ее досуха и остаток в смеси с песком подвергал прокаливанию. Долгое время благодаря малым выходам при этом способе Ф. был очень дорог; сто лет позже Ф. был найден в составе костей (Ган, 1769), когда разработан метод добычи его из их золы шведским химиком Шееле (1771). Кости и в настоящее время служат важнейшим источником для получения Ф., равно и метод Шееле в своей основе продолжает применяться (см. техническую статью). Во всех случаях получается так назыв. обыкновенный, или белый, Ф.; его называют иногда и желтым. Это слабо-желтоватое, почти бесцветное и просвечивающее твердое вещество, которое обладает запахом чеснока и по виду и консистенции напоминает воск, но при низких температурах ломко. Обыкновенный Ф. кристалличен, имеет удельный вес 1,83 при 10°, при 44,3° плавится в бесцветную жидкость с большим показателем преломления и удельным весом 1,764. При нагревании в атмосфере, лишенной кислорода, Ф. кипит при 290° под обыкновенным давлением, превращаясь в бесцветный пар, плотность которого, определенная при 515° (Митчерлих) и 1040° (Девилль и Троост), оказалась почти постоянной и именно такой, что молекулярный вес вычисляется вчетверо большим атомного, т. е. частичная формула Ф. в этих пределах температуры Р4. При высших температурах, при 1500° — 1700°, плотность оказалась убывающей (В. Мейер, Бильтц и Меншинг), так что молекулярный вес вычисляется промежуточный между Р4 и Р2; эбуллиоскопический метод (Бекман) для раствора Ф. в сероуглероде приводит точно так же к формуле Р4, как и метод криоскопический, в бензольном растворе, по Гертцу; но Патерно и Назини, пользуясь тем же растворителем и наблюдая понижение температуры замерзания, пришли к промежуточному между Р4 и Р2 молекулярному весу. Если поместить Ф. в склянку с углекислым газом, запаять и подвергнуть в темноте подогреванию на водяной бане при 40° в течение нескольких дней, то на холодных стенках сосуда получается возгон Ф. в виде крупных, блестящих и неокрашенных кристаллов. Ф. почти нерастворим в воде; мало растворим в спирте, эфире, скипидаре; легко растворим в хлористой сере, треххлористом Ф. и сероуглероде, одна весовая часть которого может растворить 17—18 вес. ч. Ф. Из растворов в сероуглероде при испарении Ф. получается в форме кристаллов — ромбические додекаэдры. Из числа особых свойств Ф. — способность светиться в темноте обратила на себя внимание со времен Бранда; даже самое название этого элемента (от φως свет и φέρω несу) объясняется этой способностью. Свечение Ф. происходит только в присутствии обыкновенного, т. е. влажного, воздуха и есть результат медленного окисления, которое при очень тонком слое, когда, например, сделана надпись на чем-либо куском Ф., прекращается после полного превращения его в смесь фосфористой и фосфорной кислот. Свечение Ф. сопровождается образованием белого дыма с запахом чеснока; этот дым объясняется летучестью Ф., содержит, между прочим, озон и очень ядовит [Законченного исследования пока еще не имеется о составе атмосферы вблизи окисляющегося Ф.]. Если на воздухе оставить куски Ф., то медленное окисление, вызывая постепенное разогревание, может окончиться и воспламенением, ввиду чего Ф. должно хранить под водой. В способности Ф. самовоспламеняться можно убедиться проще всего при помощи его сероуглеродного раствора. Если таким раствором напитать пропускную бумагу, то на воздухе происходит быстрое испарение растворителя, затем медленное окисление оставшихся на бумаге кристалликов Ф., сопровождаемое выделением белого дыма и в темноте — свечением; наконец бумага загорается. Свечение Ф. имеет место и во всяком другом газе, если к нему подмешан кислород; в чистом кислороде при 20° оно, однако, не происходит, если давление равно атмосферному или выше несколько; при уменьшении давления свечение появляется и делается наиболее ярким, когда давление понижено до 1/5 атмосферы, т. е. когда оно делается равным парциальному давлению кислорода обычного воздуха. Свечение не происходит в воздухе, содержащем сероводород, пары эфира или скипидара. Ф., как сера, углерод и проч., известен в виде нескольких аллотропических изменений. Под влиянием света или нагревания обыкновенный Ф. превращается в так наз. красный Ф. (см. Фосфор, аллотропия), который в химические взаимодействия вступает гораздо труднее обыкновенного Ф., загорается только при нагревании в воздухе до 260°. Технические применения красного Ф. вместо обыкновенного обусловливаются именно этой его инертностью, которая находится, очевидно, в связи с тем, что превращение обыкновенного Ф. в красный сопровождается выделением тепла, именно 27,3 больш. кал. на граммовый атомный вес. Кроме белого и красного Ф., известен металлический или черный Ф. (см. Фосфор, аллотропия). Обыкновенный Ф. не проводит тока; красный проводит, хотя и слабо; черный проводит лучше. По формам соединений Ф. принадлежит к группе азота; по способности к взаимодействиям он очень напоминает серу, а также углерод и кремний. Все это вполне согласуется с положением Ф. в периодической системе, где он находится в V группе, в правой, менее металлической подгруппе вместе с мышьяком и сурьмой, проявляя maximum сродства к кислороду сравнительно с этими элементами, как это имеет место и для серы сравнительно с селеном и теллуром. Очень велико сродство Ф. и к галоидам, соединения с которыми являются в тех же формах, как и кислородные соединения. Сродство к водороду очень незначительно, и прямое взаимодействие в отличие от кислорода и галоидов имеет место только с водородом в момент выделения, причем фосфористый водород PH3 получается из обыкновенного Ф. с громадной примесью свободного водорода [Такой водород, как и водород, к которому примешано немного Ф. в виде пара, горит красивым зеленым пламенем, дающим спектр с пятью яркими линиями.]. С углеродом и азотом Ф. непосредственно вовсе не соединяется. Фосфористые металлы, или фосфиды, известны для многих металлов и получаются обыкновенно из элементов при нагревании: для К, Na, Mg, Zn, Cd, Cu, sn, Pt, Ее, Ni, Со, Cr и Μn они получаются легко и очень постоянны, для Ag и Au непостоянны; для Al, Bi и Hg получаются обходным путем (Granger, 1896—98); о фосфидах кальция — см. ниже. При 250° обыкновенный Ф. разлагает водяной пар, превращаясь в фосфористую кислоту и фосфористый водород и вообще представляет собой сильный восстановитель, осаждает из растворов солей золото, серебро, медь. Если где-либо подозревается присутствие обыкновенного Ф. в свободном виде, то исследуемый материал помещают в колбу с водой и кипятят: пары воды, содержащие примесь пара Ф., окрашивают бумажку, смоченную ляписом, в черный цвет (металлическое серебро), но не изменяют цвета бумажки с свинцовой солью, как это сделал бы сероводород. О присутствии Ф. в таких парах можно судить и иным способом, а именно, соединив колбу с стеклянным холодильником и работая в темноте, замечают свечение Ф. в той части холодильника, где водяной пар в значительной мере сжижается и где, следовательно, имеет место соприкосновение с воздухом.
Галоидные соединения. Ф. исключительно, можно сказать, является трех- и пятивалентным, что особенно ясно на примере его галоидных соединений. Обыкновенный Ф. горит в хлоре, превращаясь в треххлористый, PCl3, или, в случае избытка хлора, в пятихлористый, PCl5. Удобнее всего получать PCl3, открытый Гей-Люссаком и Тенаром (1808), из красного Ф., для чего помещают его в реторту и, пропуская сухой хлор, нагревают ее осторожно; в охлажденный приемник перегоняется в этих условиях PCl3, который нуждается в очищении от примеси PCl5, для чего к перегону прибавляют некоторое количество обыкновенного Ф. и после стояния еще раз перегоняют. PCl3 — бесцветная подвижная жидкость с очень резким запахом; кипит при 76° и при —115° еще не замерзает; имеет уд. вес 1,613 при 0° и обладает плотностью пара, вполне отвечающею формуле. На воздухе дымит, реагируя с его влажностью и превращаясь в хлористый водород и фосфористую кислоту. Весьма энергично реагирует с серным ангидридом, раскисляя его:
SO3 + PCl3 = OPCl3 + SO2,
при чем получается сернистый газ и хлорокись Ф., где этот элемент уже пятивалентен. Способность быть пятивалентным, стремление к пятивалентному типу в большой мере принадлежит Ф., что очевидно и из способа получения PCl5. Это соединение открыто Деви (1810) и анализировано Дюлонгом (1816). Получают его, пропуская сухой хлор над поверхностью PCl3, находящегося в сосуде, охлаждаемом водой, до полного исчезновения жидкости — PCl3 + Cl2 = PCl5; реакция сопровождается большим выделением тепла, почему и необходимо заботиться об охлаждении. PCl5 есть белый или желтовато-белый, кристаллический порошок, обладающий очень острым неприятным запахом, который раздражает слизистую оболочку носа и вызывает слезы, и способный возгоняться, не плавясь, при 100°; при 148° и под увеличенным давлением он, однако, плавится и затем при охлаждении застывает в виде прозрачных призм. Определения плотности пара (Дюма) показали, что она ниже отвечающей приведенной формуле уже при 182°, постепенно уменьшается по мере повышения температуры и при 336° вдвое меньше вычисленной; так как вместе с тем почти не окрашенные вначале пары постепенно приобретают окраску свободного хлора, то такое непостоянство их плотности указывает на диссоциацию, именно![]() , что доказано методом диффузии, а также и путем определения плотности пара для смеси PCl5 с PCl3 (Вюрц). В совершенно сухом воздухе PCl5 не претерпевает никаких изменений, но в присутствии влажного воздуха он дымит и постепенно расплывается, превращаясь в хлорокись Ф., о которой уже упомянуто выше. Реакцию с влажностью воздуха можно представить так:
, что доказано методом диффузии, а также и путем определения плотности пара для смеси PCl5 с PCl3 (Вюрц). В совершенно сухом воздухе PCl5 не претерпевает никаких изменений, но в присутствии влажного воздуха он дымит и постепенно расплывается, превращаясь в хлорокись Ф., о которой уже упомянуто выше. Реакцию с влажностью воздуха можно представить так:
PCl5 + Н2O = HO—PCl4 + HCl = OPCl3 + 2HCl.
Такое же взаимодействие происходит, понятно, и при смешении с жидкой водой, если взять PCl5 и Н2O частица на частицу, как показал Вюрц (1847); но более удобный способ получения OPCl3 дан был Жераром, именно путем нагревания PCl5 с безводной щавелевой кислотой, которая дает вместе с тем оба окисла углерода и хлористый водород:
PCl5 + С2O4Н2 = OPCl3 + 2HCl + СО + СО2.
К числу удобных способов получения относится также окисление PCl3 бертолетовой солью, которую должно прибавлять к PCl3 малыми порциями — на 500 гр. 160 гр., пока не будет происходить вскипания (Е. Dervin):
3PCl2 + KClO3 = 3OPCl3 + KCl;
в заключение отгоняют продукт, отбрасывая первую порцию, содержащую свободный хлор. Хлорокись Ф. — бесцветная подвижная жидкость, кипящая при 107° и имеющая уд. вес 1,7118 при 0°. При сильном охлаждении она кристаллизуется и плавится затем при —1,5°. Плотность пара отвечает формуле. На воздухе дымит и походит по запаху на треххлористый Ф. Относительно PCl3 и PCl5 благодаря отношению их к воде обыкновенно говорят, что это типические хлорангидриды и именно фосфористой и фосфорной кислоты, но, как видно из дальнейшего, собственно только хлорокись Ф. есть настоящий хлорангидрид ортофосфорной кислоты. Соответствующие соединения Ф. с бромом весьма похожи и по способам получения, и по свойствам на хлористые. Жидкий трехбромистый Ф. PBr3 имеет уд. вес 2,925 при 0° и кипит при 175°. Кристаллический пятибромистый Ф. PBr5 лимонно-желтого цвета, уже при 100° превращается в красную жидкость и разлагается на PBr3 и Br2. Бромокись Ф. OPBr3 представляет плоские таблички удельного веса 2,822, плавится при 46° и кипит при 195°. С йодом Ф. дает также два соединения; но из них более богатое йодом есть трехйодистый Ф., PJ3; другое же — двуйодистый Ф., P2J4. Чтобы приготовить Р2J4, берут раствор 1 вес. части Ф. в сероуглероде и постепенно прибавляют 8,2 вес. частей йода, затем отгоняют при осторожном нагревании растворитель, после чего и остается желтая кристаллическая масса Р2J4, которая плавится при 110° и водой разлагается на йодистый водород, фосфористую кислоту и красный Ф. (1/3 всего Ф.). Оперируя таким же способом, но беря в полтора раза больше йода, получают PJ3, который представляет красные шестисторонние кристаллы, плавящиеся при 55° и водой разлагающиеся без выделения Ф. Плотность пара для Р2J4 и PJ3 была определена Троостом при 265° и незначительном давлении (59—90 миллим.) и оказалась отвечающей их формулам. Что касается фтористых соединений Ф., то они, имея состав, аналогичный хлористым, представляют по своим свойствам, как часто бывает для веществ, содержащих фтор, значительные особенности. Трехфтористый Ф. PF3 — бесцветный газ, который не только не дымит, встречаясь с обыкновенным воздухом, но и водой поглощается весьма медленно, при чем превращается в фосфористую и плавиковую кислоты. Получают PF3 (Муасан) действием фосфида меди на фтористый свинец при нагревании в медной трубке, но лучше — путем приливания по каплям трехфтористого мышьяка к PCl3, в отсутствие влажности; реакция PCl3 + AsF3 = AsCl3 + PF3 совершается энергично; газ пропускают через охлажденную до 15° трубку и промывают небольшим количеством воды и, для осушения, крепкой серной кислоты. PF3 имеет соответствующую формуле плотность; при —10° и под давлением в 40 атмосфер сгущается в подвижную неокрашенную жидкость. Пятифтористый Ф. PF5 впервые был получен (Торпе) при взаимодействии PCl5 с AsF3, именно 3PCl5 + 5AsF3 = 3PF5 + 5AsCl3; но совершенно чистый он возникает (Муасан) из продукта присоединения брома к PF3. Последний, PF3Br2, получается в виде янтарно-желтой жидкости при —10°, если действовать на сухой бром высушенным посредством крепкой серной кислоты и плавленого едкого кали трехфтористым Ф., и при —20° превращается в маленькие желтоватые кристаллики, которые по вынимании сосуда из охладительной смеси тотчас плавятся. Это вещество, сохраняемое в запаянной трубке, уже при 15° постепенно разлагается на кристаллический PBr5 и газообразный PF5:
5PF3Br2 = 3PF5 + 2PBr5;
как будто снова кристаллизуется при обыкновенной температуре в виде желтых кристаллов. Получающийся из PF3Br2 газ содержит только некоторое количество паров брома и легко может быть очищен посредством ртути, над которой его можно собрать и некоторое время сохранять, после чего он постепенно начинает уменьшаться в объеме, действуя на стекло и проч. PF5 есть бесцветный газ с резким запахом, сильно действует на слизистые оболочки, водой разлагается на фосфорную и плавиковую кислоты. В аппарате Кальете он сжижается при 16° под давлением 46 атмосфер в жидкость, не действующую на стекло, которая при быстром понижении давления замерзает в снегоподобную массу. Плотность PF5 вполне соответствует формуле; частица его очень прочна, что видно из безразличного его отношения к ряду искр от индукционной катушки не только в чистом виде, но и в смеси с водородом или кислородом (Торпе); только действие сильной спирали вызывает его распадение (Муасан) на PF3 и F2, который соединяется тогда со ртутью, причем объем газа не меняется. Фторокись Ф. OPF3 получается (Муасан) из смеси PF3 с половинным объемом кислорода при взрыве от электрической искры или при пропускании над нагретой губчатой платиной. Это газ, который сжижается при —50° или под давлением 15 атм. при 16° и замерзает при быстром уменьшении давления; он дымит на воздухе и разлагается водой, что само собой понятно из приведенного уже отношения PF5 к воде. О галоидных соединениях Ф. можно вообще сказать, что те из них, которые содержат трехвалентный Ф., охотно присоединяют галоид, переходя в высший тип пятивалентного Ф., причем могут получаться и смешанные соединения, напр. PF3Cl2, PCl3Br2 и проч., а также и кислород, действуя иной раз как восстановители. Но и высшего типа галоидные соединения не лишены способности присоединять, вступая в так называемые молекулярные соединения. PCl5 соединяется со многими хлористыми металлами, напр. FePCl8, и некоторыми неметаллами, напр. с хлористым селеном, йодом — SeCl4∙PCl5 и PCl5∙ClJ. С бромом PCl3 дает не только PCl3Br2, но и PCl3Br2∙Br2 и PCl3Br2∙2Br2. Некоторым из галоидных соединений Ф. принадлежит способность присоединять аммиак; напр. когда пропускают газообразный NH3 в раствор PCl5 в четыреххлористом углероде, то получается (Besson) в осадке белый порошок состава PCl5∙8NH3, a для PF5 известно желтоватое твердое соединение 2PF5∙5NHЗ. В лабораториях галоидные соединения Ф. находят многочисленные применения, особенно в области органической химии. PCl3 превращает кислоты в хлорангидриды, при чем получается фосфористая кислота [Возможны вспышки, если работают при нагревании более сильном, чем на водяной бане, так как фосфористая кислота способна разлагаться при выделении фосфористого водорода, легко загорающегося в горячем состоянии.]; то же делает и PCl5, но при образовании хлорокиси Ф. Кроме того, PCl5 способен выменивать два хлора на кислород альдегидов или кетонов, превращая их в соответствующие двухлоропродукты, а также он способен и хлорировать, т. е. производить металептическое замещение водорода органических соединений, при чем превращается в PCl3; такое хлорирование в струе свободного хлора происходит и в присутствии малых количеств PCl5; возникающий PCl3 все время превращается хлором в PCl5; как хлористая сурьма, PCl3 может быть, следовательно, передатчиком хлора. Хлорангидриды кислот из солей получаются при нагревании с OPCl3, которая при этом превращается в фосфат. Для приготовления газообразного йодистого водорода готовят PJ3 и приливают по каплям воду; при получении газообразного бромистого водорода действуют бромом на красный Ф. в присутствии малого количества воды, что сводится к реакции ее с PBr3; в обоих случаях получается, очевидно, еще и фосфористая кислота. PBr3 и PJ3 широко применяются также для получения бромистых и йодистых органических радикалов из различных спиртов.
Кислородные соединения. Когда Ф. горит в избытке сухого воздуха, то в отличие от серы образуется высшая степень окисления, где Ф. пятивалентен, как и в PCl5 или OPCl3; это фосфорный ангидрид Р2О5, аналог азотного. На граммовую частицу тепла выделяется при этом около 370 больш. калорий — громадное количество, превышающее теплоту образования эквивалентного веса угольного ангидрида раза в полтора, а жидкой воды на 15 %. Так как Р2О5 имеет большое сродство к воде, то для сжигания Ф. — это единственный путь получения Р2О5 — употребляют различные аппараты, имеющие целью или полное высушивание потребного для горения воздуха, или, по крайней мере, предохранение продукта от влажности окружающего воздуха, и помещают его в хорошо закрывающиеся сосуды, напр. стеклянные банки с притертой и залитой парафином пробкой. Горение совершается с ярким пламенем и обильным выделением белого дыма, который медленно осаждается на стенках и дне аппарата и представляет искомый продукт, легкое, белое, хлопьевидное вещество. Аппарат Грабовского служит для получения значительных количеств Р2О5 — это цилиндр а из листового железа, 35 см высоты и 30 ширины; он закрыт крышкой с загнутой трубкой в 25 мм ширины, просвет коей может быть изменяем посредством срезанной пробки с; дном цилиндра служит железная воронка h, которая вставлена в склянку g; просвет между h и нижним краем a можно регулировать толщиной подставки i.

Сухой Ф. помещают на нагретую медную с железной ручкой ложку d и вносят в цилиндр, закрепляя ее в е. Воздух обменивается через упомянутый просвет и трубку b. Около 0,7 продукта попадает при этом в склянку, где тотчас и закрывается пробкой. Обыкновенный Р2O состоит главным образом из аморфного порошка, отчасти из кристаллического. Кристаллическое видоизменение может быть получено (Hautefeuille и Perreg) путем возгонки обыкновенного Р2O5 при осторожном нагревании не выше 250° в токе углекислого газа и состоит из отдельных моноклиномерных кристалликов, которые не окрашены, прозрачны и обладают большим показателем преломления, или из их хлопьевидных скоплений. Упругость пара кристаллического P2O5 при 250° равна 760 миллиметрам; при немного высшей температуре она падает до нескольких только миллиметров — происходит, по всей вероятности, полимеризация [Плотность пара Р2O5, определенная по способу В. Мейера при ярко-красном калении (Fildun и Barnett, 1896), дала молекулярный вес 326, что значительно выше 284, величины, отвечающей Р4О10, и ниже 426 — для Р6О15; вероятно, в парах присутствует смесь Р2О5 и (Р7О5)n.] и превращение в аморфное видоизменение, это превращение идет уже при 300° и легко при 440°; оно экзотермично, как показывает меньшая теплота растворения аморфного Р2O5 сравнительно с кристаллическим. Растворение в воде кристаллического Р2O5 совершается быстро — сразу получается прозрачный раствор; аморфный же дает с водой сначала студенистые комки, которые довольно долго не растворяются. При красном калении оба видоизменения плавятся в прозрачную жидкость, которая при охлаждении застывает в стекловатую массу — стекловидное видоизменение Р2O5, растрескивающуюся по всем направлениям со слабым свечением и звуком. Такой Р2O5 в воде растворяется очень медленно и медленно же, но вполне, превращается при возгонке в кристаллический. Когда вносят Р2O5 в воду, то происходит растворение, сопровождаемое шипением, как будто от раскаленного железа; таково сродство ангидрида к воде [Благодаря такому отношению к воде Р2O5 является мощным высушивающим средством как для газов, так и для жидкостей, с ним не реагирующих, а также и для твердых тел — в эксикаторе. Р2O5 применяется, кроме того, для получения ангидридов из кислот, например азотного, серного.]. Однако полное насыщение Р2O5 водой требует времени и нагревания на водяной бане, потому что только тогда в растворе возникает нормальный гидрат, так называемая ортофосфорная кислота, частица которой содержит соединенные с атомом Ф. четыре атома кислорода, как серная и другие ортокислоты, и три водорода, как в высшем водородистом соединении Ф., что аналогично с нахождением в частице серной кислоты двух водородов, а в частице хлорной — одного. Все превращение выражается так:
Р2O5 + 3Н2О = 2HЗPO4.
Та же самая трехосновная кислота получается и при взаимодействии хлорокиси Ф. с водой — реакция, позволяющая заключить самым очевидным образом о присутствии в ортокислоте трех водных остатков, о чем уже упомянуто выше. Возможность получить ортокислоту из PCl5 сама собой очевидна, так как первый продукт его взаимодействия с водой есть Cl3РО; но обычный путь получения чистой ортокислоты состоит в окислении красного Ф. крепкой азотной кислотой в реторте при подогревании; происходит выделение бурых паров (азотная кислота раскисляется) и постепенное растворение Ф.; полученный раствор сгущают в фарфоровой чашке, подбавляя время от времени крепкой азотной кислоты, чтобы не осталось фосфористой кислоты; удаляют окислы азота, нагревая не выше 188°; обрабатывают сероводородом для удаления мышьяка, который обыкновенно присутствует в продажном Ф. [Этим именно объясняется (Cl. Winkler) недавнее «превращение» Ф. в мышьяк (F. Fittica, 1900), который попадает в Ф. из серной кислоты, употребляемой для обработки костей.], несколько разбавляют водой, фильтруют и снова выпаривают не выше, как при 160°. К той же цели приводит и окисление обыкновенного Ф.; но азотную кислоту во избежание вспышки приходится брать разведенную, удельного веса 1,2, что замедляет операцию. Из костяной золы фосфорная кислота получается различными способами. Например, по Либиху, обливают золу равным весом серной кислоты, разбавив ее предварительно в десять раз большим количеством воды, и подвергают продолжительному кипячению; затем фильтруют через холст и фильтрат сильно сгущают; после того, прибавляя крепкой серной кислоты, осаждают не выделенный еще кальций в виде гипса и сливают прозрачный раствор, который, чтобы освободить от избытка серной кислоты, подвергают сильному сгущению вплоть до прокаливания, что приводит к полному удалению серной кислоты и превращению присутствующей магниевой соли в нерастворимый метафосфат (см. ниже). Извлечение водой таким образом полученного продукта и нагревание слитой жидкости приводит, наконец, к раствору ортокислоты. Η3PΟ4 растворима в воде во всех пропорциях. Имеет приятный, кислый вкус и лишена всякого запаха. Когда при сгущении раствора достигается состав, отвечающий формуле, ортокислота имеет вид бесцветного сиропа, который не изменяется при нагревании, если температура не превосходит 160°, и при стоянии постепенно превращается в кристаллическую массу; кристаллизация идет быстро, если бросить кристаллик в очень крепкий раствор; кристаллы принадлежат к ромбической системе (призмы) и плавятся при 38,6°. Вот таблица удельных весов при 15°/4° для водных растворов кислоты, по данным Кольрауша (см. Менделеев, «Исследов. водных растворов по уд. весу»):
| % Η3ΟΡ4 | Уд. вес. | % Н3РО4 | Уд. вес. |
|---|---|---|---|
| 10 | 1,054 | 40 | 1,254 |
| 20 | 1,113 | 50 | 1,330 |
| 30 | 1,180 | 60 | 1,427 |
Удельный вес самой кислоты, переохлажденной, 1,88 при 18°. Если на OPCl3 действовать обыкновенным спиртом, то в зависимости от относительных количеств и условий взаимодействия происходят следующие реакции:
1) OPCl3 + 3C2H5—OH = OP(O—C2H5)(OH)2 + 2C2H5—Cl + HCl;
2) OPCl3 + 3C2H5—ОН = ОР(O—C2H5)2—ОН + C2H5—Cl + 2HCl;
3) OPCJ3 + 3C2H5—ОН = OP(O—C2H5)3 + 3HCl;
получаются одноосновная и двухосновная эфирокислоты — сиропообразные, непахучие и бесцветные жидкости, дающие кристаллические соли, — и эфир ортофосфорной кислоты, который кипит при 215°, имеет удельный вес 1,072 при 12° и может быть получен, как обыкновенно для сложных эфиров, также при действии йодистого этила на Ag3PO4. При нагревании ортокислоты выше 160° она начинает постепенно терять воду. Если нагревание не превосходит 213°, то процесс ограничивается потерей одной частицы воды из двух частиц кислоты:
2HЗPO4 = Н4P2О7 + Н2O;
возникает одна частица новой, четырехосновной кислоты. Это пирофосфорная кислота O:(PO2)2(OH)4 [Возможно существование и более сложных кислот этого рода, как, напр., недавно полученная (Schwarz, 1895) пятиосновная пирокислота О2(РО)3(ОН)5. Медная соль ее была приготовлена из продукта сплавления частичных количеств пиро- и метафосфатов натрия и при содействии сероводорода превращена в свободную кислоту, которая, подобно метафосфорной, свертывает белок и в виде натриевой соли не дает осадков с солями Со, Ni, Cu и Zn — осаждает только в виде очень крепких, да и то не вполне — чем отличается от пирокислоты обыкновенной. Отвечающий последней кислоте хлорангидрид, хлористый пирофосфорил, O(PO2)2Cl4, получается при окислении PCl3 азотноватым ангидридом; это жидкость, которая кипит, не без разложения, при 210—215°.], представляющая собой то мягкую стекловатую, то полупрозрачную неясно-кристаллическую массу. Для ортокислоты известны три ряда солей; например, для натрия: однометаллическая ортофосфорно-натриевая соль, или дигидроортофосфат натрия NaH2PO4, двуметаллическая, или гидроортофосфат Na2HPO4, и трехметаллическая или ортофосфат Na3PO4. Ортофосфаты щелочных металлов, за исключением литиевого, хорошо растворимы в воде и имеют щелочную реакцию; реакция гидроортофосфатов средняя, а для дигидроортофосфатов кислая; те и другие для щелочных металлов в воде растворимы. Орто- и гидроортофосфаты прочих металлов в воде обыкновенно нерастворимы, например Са3(РО4)2, FePO4, MgHPO4, но растворяются в кислотах, превращаясь в дигидроортофосфаты, которые почти всегда растворимы, напр. Са(Н2РО4)2. Для пирокислоты мыслимы четыре рода солей, но известны только два [Для натрия недавно получена еще и такая соль — Na3HP2O7∙H2O, соль же NaH3P2O7 не удалось очистить (Salzer, 1894).], а именно для натрия, напр. двуметаллическая, или дигидропирофосфат Na2H2P2O7, и четырехметаллическая, или пирофосфат Na4P2O7. Последняя соль очень просто может быть получена из гидроортофосфата натрия Na2HPO4∙12Н2О, который при нагревании теряет сначала кристаллизационную воду, а затем при слабом калении — и конституционную:
2Na2HPO4 = Na4PO7 + H2O.
Из раствора этого пирофосфата проще всего получить раствор чистой кислоты, для чего, прибавляя уксусно-свинцовой соли, получают осадок пирофосфата свинца Pb2P2O7, промывают его и, разболтав с водой, разлагают сероводородом:
Pb2P2O7 + 2H2S = Н4P2O7 + 2PbS;
фильтруют от PbS и, оставляя фильтрат в плоском сосуде, избавляются от избыточного сероводорода. В виде раствора пирокислота при обыкновенной температуре остается без изменений целые месяцы; превращение в ортокислоту совершается через некоторое время при нагревании и быстро, если сверх того присутствует сильная кислота, при чем, очевидно, совершается гидролиз:
Н4P2O7 + H2O = 2H3PO4.
Отличить орто- и пирокислоту в растворе очень просто; по нейтрализации аммиаком из раствора ортокислоты ляписом осаждают ее в виде желтого ортофосфата серебра Ag3PO4, a из раствора пирокислоты получают осадок белого пирофосфата серебра Ag4P2O7; из пересыщенного аммиаком раствора ортокислоты сульфат магния осаждает неокрашенный кристаллический ортофосфат такого состава — Mg(NH4)PO4, и из подкисленного азотной кислотой раствора ее молибдат аммония осаждает желтый осадок фосфорно-молибденовокислого аммония (NH4)3PO4∙nMnO3 (см. Молибден), а с пирокислотой эти реактивы осадков не дают; при нейтрализации содой из ортокислоты получается Na2HPO4, а из пирокислоты Na4P2O7. Последняя соль есть наиболее обыкновенная из пирофосфатов; она, подобно прочим солям щелочных металлов, хорошо растворима в воде; пирофосфаты же других металлов в воде нерастворимы, но растворяются в кислотах, переходя при этом в растворимые дигидропирофосфаты. Нерастворимые пирофосфаты готовятся путем осаждения соляных растворов раствором Na4P2O7; они обыкновенно растворимы в избытке реактива — превращаются в двойные растворимые соли. Натриевая соль, получаемая при накаливании из Na2HPO4, весьма постоянна и в растворе; растворы ее выдерживают продолжительное кипячение, даже по прибавлении едкого натра; но кипячение с сильными кислотами обусловливает превращение пирокислоты в ортокислоту [По опытам Вертело и Андре (1897), при стоянии водного раствора чистой пирокислоты 9 % ее обратились в ортокислоту через два дня, a 52 % — через три месяца: в общем превращение совершалось подобно наблюденному для метакислоты.]. От описанных кислот Ф. существенно отличается та кислота, которая возникает в водном растворе в момент растворения фосфорного ангидрида, при чем получается кислая жидкость, которая с ляписом дает белый осадок, подобно пирокислоте, но в то же время проявляет способность свертывать, осаждать разбавленный белок куриного яйца, к чему пирокислота, как и ортокислота, совершенно неспособна в отличие от азотной и других минеральных кислот; к тому же и состав нерастворимой серебряной соли AgPO3, как и солей прочих металлов, ясно говорит за ее индивидуальность. Это — метафосфорная кислота НРО3, которая одноосновна, как азотная, и образуется, очевидно, через присоединение к Р2О5 одной частицы воды, между тем с двумя частицами воды должна бы получаться пирокислота, а с тремя возникает ортокислота:
Р2О5 + H2O = 2HPO3
Р2О5 + 2H2O = Н4Р2О7
Р2О5 + 3H2O = 2Н3РΟ4.
Первая частица воды присоединяется тотчас по смешении P2O5 с водой, но чтоб произошла ортокислота, требуется время и нагревание даже [По опытам Сабатье (1888—89) в более крепких растворах НРО3 быстрее, чем в слабых, превращается в Н3РО4; полунормальный раствор для этого требует при 0° до 150 дней, при 31° — около 5 дней, а при 95° менее одного часа. Присутствие сильных кислот ускоряет (указано и Греемом) превращение, уксусная кислота задерживает, как это имеет место и для нейтрализованной сильным основанием метакислоты. Образование при этом процессе также и пирокислоты наблюдается (Бертело и Андре, 1897) в ничтожном количестве.], о чем уже было упомянуто. Когда нагревают ортокислоту с целью получить пирокислоту, т. е. не выше, чем при 213°, то продукт обыкновенно содержит примесь мета- и ортокислоты, которые в сумме по составу равны пирокислоте. Более сильное нагревание, прокаливание, — вызывающее плавление и даже испарение — приводит к полному превращению ортокислоты в метакислоту. При охлаждении расплавленная НРО3 застывает в густую тестообразную массу, расплывающуюся на воздухе; если же было подбавлено немного пирофосфата натрия, то — в бесцветное стекло, и известна тогда под именем стекловидной или ледяной фосфорной кислоты — Acidum phosphoricum glaciale [Подбавляют к ней фосфата натрия или соды для связности и твердости. Открыть количество натрия очень просто: из раствора такой НРО3 крепкая соляная кислота осаждает его вполне в виде NaCl (Bettendorff).], находящейся в продаже в форме палочек, которые хорошо растворимы в воде и кажутся во время процесса растворения ледяными или стеклянными. История установки существования описанных трех кислот, ангидрид коих один и тот же, представляет большой интерес, так как при этом были констатированы факты, положенные (Либих) в основу теории многоосновных кислот в связи с теорией водородных кислот. И серный ангидрид, кроме нормального гидрата, Н2SO4, образует другие гидраты — Н4SO5, H6SO6 и проч., но химические превращения серной кислоты почти всегда, особенно в растворах, идут на счет нормального гидрата. Гидраты Р2О5 необыкновенно устойчивы и являются даже самостоятельными кислотами. В 1746 г. Маргграф показал, что аммонийно-натриевый гидрофосфат (Sal urinae fixum sive microcosmicum) (NH4)NaHPO4∙4H2O с раствором ляписа дает желтый осадок серебряной соли. В 1828 г. Кларк нашел, что гидрофосфат натрия или обыкновенная фосфорно-натриевая соль Na2HPO4∙12H2O, которая дает такую же серебряную соль, после прокаливания с ляписом выделяет белый осадок; таким образом было констатировано существование пирокислоты. В 1830 г. Энгельгардт нашел, что мнение о неспособности фосфорной кислоты свертывать белок несправедливо, ибо раствор фосфорной кислоты, приготовленный из безводной кислоты (ангидрида), свертывает, хотя затем и теряет эту способность. Эти и подобные наблюдения повели к допущению, что фосфорная кислота, или, по-теперешнему, ангидрид, способна изомеризоваться. Истинная причина всех этих явлений установлена была, наконец, Греемом в 1833 г., который обратил внимание на различный состав солей и гидратов кислотных, а вместе с тем установил и существование метакислоты [Хлорангидрид ее образуется вместе с хлористым пирофосфорилом при нагревании с небольшим количеством воды (Besson, 1897); Cl3PO + Η2O = ClPO2 + 2HCl и 2Cl3PO + Η2O = Cl4P2O3 + 2HCl; кроме того, получается и H3PO4. При 110° под давлением в 10 мм для Cl4P2O3 имеет место такое разложение: Cl4P2O3 = Cl3PO + ClPO2.]. Для Р2О5, однако, действительно известно несколько видоизменений благодаря, по всей вероятности, его способности к полимеризации; после присоединения к Р2О5 воды возникает метакислота, для которой также существуют, и притом многие, видоизменения одного и того же состава (НРО3)n — полиметафосфорные кислоты, известные, впрочем, почти исключительно в форме солей — (МРО3)n, где M — одновалентный атом металла, аммоний также. О степени полимерности метафосфатов судят с некоторой вероятностью по составу двойных солей (Fleitmann и Henneberg), а также применяя метод Рауля и изучая электропроводность растворов (Тамман, 1890 и 1892). Исследованы главным образом метафосфаты, получаемые из NH4HPO4 и из различных дигидроортофосфатов 1) путем осторожного нагревания, пока не окончится выделение воды (и аммиака) и не будет достигнута только слабая кислая реакция, 2) при более сильном нагревании — 350° и 316°, а также 3) прокаливании до сплавления; в последнем случае быстрое охлаждение дает соли более полимерные, чем медленное, когда есть время для кристаллизации. Диметафосфат натрия Na2(РО3)2∙4Н2O образуется из (NH4)NaHPO4 и именно при возможно слабом нагревании вместе с нерастворимым метафосфатом; его извлекают водой и кристаллизуют путем испарения в теплом месте (30°) или осаждают спиртом; последний прием позволяет достигнуть полной чистоты соли (Тамман). Через двойное разложение получаются из этой соли другие диметафосфаты: К2(РО3)2, Ba(PO3)2∙2Н2O, 3Ag2(PO3)2∙H2O и проч. Вообще соли диметакислоты растворимы, не исключая и серебряной, что уподобляет ее азотной кислоте; но с уксуснокислым свинцом получается осадок. При обыкновенной температуре растворы Na2(PO3)2 не изменяются и нейтральны; при кипячении появляется кислая реакция вследствие возникновения NaH2PO4. При довольно сильном нагревании (316° и 350°) Н3PO4 с окисями или с солями, если только они содержат летучие кислоты, кобальта, цинка, меди, марганца получаются нерастворимые триметафосфаты этих металлов; окиси более тяжелых металлов (Pb, Bi, Cd) в тех же условиях дают гексаметафосфаты. Триметафосфаты Na, К, NH4 образуются при действии растворов соответствующих сульфидов M2S на триметафосфат меди. Na3(PO3)3∙3H2O есть соль хорошо растворимая в воде и очень склонна образовать двойные соли. До исследований Таммана триметафосфаты считались диметафосфатами, и обратно. Когда подвергают разложению Na(NH4)HPO4 или NaH2PO4 и затем сплавлению или когда плавят вообще какой-либо метафосфат натрия, то при быстром охлаждении получается стекловидный, расплывчатый на воздухе и, следовательно, растворимый в воде продукт; это гексаметафосфат натрия Na6(PO3)6, или соль Греема, обыкновеннейшая из метафосфатов, выражаемая приведенной формулой уже давно (Fleitmann, 1849), так как с нашатырем из нее была получена такая двойная соль (NH4)5Na(POЗ)6. Новейшие исследования (Тамман) подтвердили состав частицы соли Греема и, кроме того, показали, что она содержит в виде примеси еще две соли, которые также гексаметафосфаты, но изомерные или, быть может, полимерные, и могут быть выражены таким образом — Na5[Na(PO3)6] и Na4[Na2(PO3)6]: при взаимодействии соли Греема с избытком ляписа получается кристаллический осадок Ag6(PO3)6 и смолообразная, легко отделимая механически от Ag6(PO3)6 смесь двух солей — Ag5[Na(PO3)6] и Ag4[Na2(PO3)6]; т. е. соль Греема содержит некоторое изменчивое в зависимости от скорости охлаждения количество метафосфатов, коих металлические атомы не все вымениваются при двойном разложении. Подвергнув обработке раствором поваренной соли чистый Ag6(PO3)6, получают чистый же Na6(PO3)6, который весьма легко растворим в воде и по химическим отношениям очень близок к соли Греема, представляя некоторые отличия благодаря отсутствию указанных примесей: обе соли дают с соляными растворами металлов, за исключением металлов щелочей, осадки, растворимые или в избытке реактива, или в избытке метафосфата; только с хлористым барием и уксуснокислым свинцом получаются не растворимые ни в том, ни в другом реактиве осадки. Единственное почти отличие чистого Na6(PO3)6 от соли Греема в том, что осадок с AgNO3, а также и с SrCl2, для первой клочьевидный, а для второй легко собирается при взбалтывании в комья. Для щелочных металлов, как и для аммония, существуют еще нерастворимые или очень трудно растворимые метафосфаты; до последнего времени им придавали простейшую формулу; это были монометафосфаты МРО3, хотя естественнее было бы полагать, что они, как нерастворимые, полимернее описанных метафосфатов. Согласно новейшим исследованиям (Тамман), это так и обстоит. Метафосфат аммония, который получается (Fleitmann) при 200° — 250° из растворимой и хорошо кристаллизующейся соли (NH4PO3)3 [По Флейтману, это (NH4)2(PO3)3.] — без изменения веса, но с потерей прозрачности и способности растворяться, вероятно, имеет такую частицу (ΝΗ4)10(ΡΟ3)10, так как через двойное разложение из него получены соли: Sr5(РО3)10∙13Н2O, Mn5(РО3)10∙12Н2O, Pb5(РО3)10∙6Н2O, Ag10(РО3)10∙8Н2O, а также K9(NH4)(РО3)10∙10Н2O. В горячей воде (ΝΗ4)10(ΡΟ3)10 растворяется, распадаясь, так как в полученном растворе оказывается уже (NH4)5(ΡΟ3)5 — соль, хорошо растворимая в воде и с хлористым калием, через двойное разложение, превращающаяся в двойную соль ΝΗ4[Κ4(ΡΟ3)5]∙6Н2O. Эта двойная соль кристаллизуется в мельчайших шестисторонних табличках, а соли, полученные с помощью NaBr и LiBr — NH4[Na4(PO3)5] и NH4[Li4(PO3)5] — полужидки. Из нерастворимого метафосфата натрия, который возникает вместе с Na2(PO3)2 при осторожном нагревании (NH4)NaHPO4, с раствором эквивалентного количества ляписа получается Na11Ag(PO3)12, а с соответствующим количеством хлористого калия — Na11K(PO3)12. Нерастворимый метафосфат натрия, образующийся вместе с солью Греема, оказался еще более сложным. Что касается кислот во всех этих метафосфатах, то они, подобно обыкновенной метакислоте, обладают способностью свертывать белок и ближе пока не характеризованы. Метакислота обычная, т. е. приготовленная нагреванием и сплавлением ортокислоты, не представляет чистой гексакислоты (α), отвечающей, как обыкновенно думают, соли Греема, а содержит и другие метакислоты (Тамман), по крайней мере одну еще (β). Так как β-кислота находится не только в водном растворе обыкновенной метакислоты, но присутствует и в растворах P2О5, то происхождение ее из ортокислоты находится, вероятно, в связи с некоторым разложением расплавленной НРО3 на воду и ангидрид, который, как малолетучий, и остается в виде примеси к охлажденному продукту (Тамман). Соли β-кислоты для К, Na и (NH4) кристалличны, вследствие чего и могут быть отделены от солей α-кислоты.
Как из OPCl3 при действии воды получается фосфорная кислота Η3PΟ4, так из PCl3 — фосфористая кислота Н3PO3. Нет надобности пользоваться для этого готовым PCl3; берут обыкновенный Ф., расплавляют его в колбе под водой и вводят хлор. PCl3 по мере возникновения превращается водой в искомый продукт и хлористый водород:
PCl3 + 3Н2O = Н3PO3 + 3HCl,
которые и растворяются. Как только исчез почти весь Ф., прекращают пропускание хлора и полученный раствор выпаривают, пока остаток не достигнет температуры в 180° — это чистая фосфористая кислота, имеющая вид густого бесцветного сиропа, который при охлаждении более или менее быстро превращается в кристаллическую массу, плавящуюся при 70°. H3PO3 имеет кислый с чесночным оттенком вкус; растворима во всякой пропорции в воде и на воздухе расплывается. Согласно с фактом получения H3PO3 из PCl3 можно было бы полагать, что это трехосновная кислота, подобно фосфорной. В действительности она оказывается двухосновной и образует только два ряда солей — фосфиты М2(НРО3) и гидрофосфиты МН(HPO3). Соли щелочных металлов растворимы в воде; соли прочих металлов по большей части нерастворимы, как, например, кальциевая, бариевая, свинцовая. Двухосновность Η3PΟ3 довольно давно уже объяснялась присутствием в ее частице только двух водных остатков, а не трех; можно было полагать, что продукт взаимодействия PCl3 с водой в момент образования превращается вследствие стремления Ф. окисляться и вообще быть пятивалентным в такую двухосновную кислоту — Р(ОН)3 = Н—РО(ОН)2. Такое предположение доказано с полной очевидностью в последние годы (A. Michaelis и Th. Becker, 1897): если подействовать при нагревании (175° в течение 60 часов) на фосфит свинца йодистым этилом, то получается соответствующий сложный эфир (темп. кип. 184° — 185°): H—PO(O2Pb) + 2C2H5J = H—PO(O—C2H5)2 + PbJ2; это соединение, растворенное в обыкновенном эфире, дает кристаллическое натриевое производное при осторожном прибавлении металлического натрия, что совершается при выделении водорода и требует для полного завершения продолжительного нагревания на водяной бане с обратным холодильником; затем охлаждают, прибавляют C2H5J и снова подвергают нагреванию:
Na—PO(O—C2H5)2 + C2H5J = C2H5—PO(O—C2H5)2 + NaJ;
после нового охлаждения эфирный раствор отфильтровывают от йодистого натрия, фильтрат избавляют от растворителя испарением и очищают остаток перегонкой в атмосфере углекислого газа. Из свинцовой соли, а следовательно, из обычной Н3PO3 получается таким образом этиловый эфир — бесцветная жидкость удельного веса 1,025 при 21° и с температурой кипения 198° — этилфосфиновой кислоты (см. Фосфины), который только изомерен с этилфосфитом, получаемым при взаимодействии ΡCl3 с алкоголятом натрия (PCl3 + 3C2H6—O—Na = [C2H5—O]3P + 3NaCl) и представляющим бесцветную жидкость удельного веса 1,075 при 15° с темп. кип. 191°. Сложный эфир из свинцовой соли Н3PO3 при омылении дает этилфосфиновую кислоту, которая содержит этил в непосредственном соединении с атомом Ф.; этиловый же эфир из PCl3 распадается при омылении на спирт и обыкновенную Н3PO3. Трехосновная фосфористая кислота существует, следовательно, только в форме эфиров, обычная же двухосновна; водород ее, непосредственно связанный с Ф., при действии едких щелочей не замещается металлом, как и водород в РН3 [1]. Аналогичные отношения имеют место и для сернистой кислоты H—SO—OH (см. Сера), но здесь оба водорода реагируют со щелочью — на том основании, что и H2S проявляет свойства кислоты. Одновалентный остаток ортофосфорной кислоты, или фосфорилдигидроксил РО(ОН)2, аналогичен, следовательно, с таковыми же сульфоксилом SO2(OH) и карбоксилом СО(ОН). В виде водных растворов Н3PO3 медленно поглощает кислород воздуха и способна восстановлять золото, серебро (получается черный осадок металла при действии ляписа), ртуть из их соляных растворов, превращаясь в Η3PΟ4; она восстановляет при кипячения даже сернистую кислоту — до серы, но водородом в момент выделения сама восстановляется, превращаясь в РН3. При довольно сильном нагревании (200°) и в отсутствие воздуха, чтобы не было окисления, Н3PO3 распадается так:
4Н3PO3 = 3Η3PΟ4 + РН3.
Соли Н3PO3 разделяют с ней восстановительные свойства. Отвечающим Н3PO3 окислом, и именно как SO2 отвечает сернистой кислоте, является так назыв. фосфористый ангидрид, аналогичный по частичной формуле мышьяковистому, т. е. это не Р2О3, а Р4О6. Он получается при сжигании Ф. в медленной струе воздуха, при чем возникает еще и Р2О5, а также недокись Р4О и, быть может, закись Р2О. Если воздух из трубки с окисляемым им обыкновенным Ф. или, лучше сказать, азот этого воздуха проходит через латунную трубку, нагретую до 50° — 60° и содержащую пробку из стеклянной ваты, то труднолетучий Р2О5 остается в этой трубке, а Р4О6 гонится далее и сгущается в U-образной трубке, помещенной в охладительной смеси; низшие окислы остаются там, где образовались (Thorpe и Tutton, 1890). Р4О6 получается в виде воскоподобной массы или перистых кристаллов, а при охлаждении из расплавленного состояния превращается в тонкие, вероятно, моноклиномерные призмы; плавится он при 22,5° и имеет удельный вес 1,9358 при 24,8°; кипит при 173° в атмосфере азота и имеет плотность пара, отвечающую приведенной формуле, которая согласуется с понижением температуры замерзания бензольного раствора. Холодная вода действует на P4O6 медленно, превращая его в Η3PΟ3, с горячей же водой происходит бурная реакция, сопровождающаяся образованием Η3PΟ4, РН3, красного Ф. и проч. На воздухе P4O6 окисляется самопроизвольно в Р2O5 и при 50—60° воспламеняется; горит также и в хлоре. Растворим без изменения в эфире, хлороформе, бензоле, сероуглероде, но со спиртом реагирует энергично, превращаясь в сложный эфир, который кипит при 184—185°, имеет удельный вес 1,075 при 15,5° и слабокислую реакцию, вследствие чего может считаться эфиром трехосновной фосфористой кислоты — НО—Р(O—C2H5)2. При нагревании в запаянной трубке мутится уже при 210°, а при 440° нацело распадается на красный Ф. и новый окисел, промежуточный между Р2O5 и P4O6; это — фосфорноватая окись Р2О4 [Этот окисел был приготовлен и ранее, чем P4O6 (Thorpe и Fulton, 1887); частичный вес его не определен. Неизвестны частичные веса и для упомянутых Р4О и P2O.]. Подобно азотноватой окиси Р2О4 охотно растворяется в воде с большим выделением тепла и не представляет собой самостоятельного ангидрида: в растворе присутствуют две кислоты — Н3РО4 и Η3PΟ3. В запаянной трубке Р2О4 в атмосфере СО2 может быть возгнана в виде прозрачных блестящих кристаллов, которые при 100° еще не плавятся, а при 180° улетучиваются, образуя такой же возгон. Кристаллы Р2О4 расплывчаты на воздухе. Существует, однако, и кислота, отвечающая ангидриду состава Р2О4, — четырехосновная фосфорноватая кислота Н4Р2О6, которая получается (Salzer) вместе с Н3PO3 и Η3PΟ4, когда обыкновенный Ф. подвергается медленному окислению влажным воздухом в ограниченном пространстве [Прежде думали, что в этих условиях образуется только Н3PO4 и Η3PO3 в изменчивых относительных количествах.]. Водный раствор продуктов такого окисления нейтрализуют едким натром и получают осадок двуметаллической соли Na2H2P2O6, которая трудно растворима. При действии сероводорода на разболтанную свинцовую соль Pb2P2O6 образуется водный раствор Η4P2O6, который может быть сгущен до некоторого предела без разложения кислоты. Раствор, полученный разложением бариевой соли, ВаН2P2О6∙2Н2O, при действии потребного количества серной кислоты и сгущенный до густоты сиропа способен кристаллизоваться (A. Joly), выделяет таблицы состава Η4P2O6∙2H2O, которые теряют воду в пустоте над крепкой серной кислотой. Чистая Η4P2O6 плавится при 55°, на холоде кристаллизуется, а при 70° распадается с выделением тепла на Η3PO3 и НРО3. От Н3PO4 отличается эта кислота неспособностью осаждаться в азотнокислом растворе и при обыкновенной температуре молибдатом аммония, а от Η3PO3 — меньшей склонностью окисляться: Ag4Р2O6 — белый осадок, получающийся при действии раствора ляписа и трудно растворимый в кислотах, не чернеет на свету даже при кипячении; хамелеон превращает, однако, Η4P2O6 в кислом растворе в Н3PO4, как — при кипячении — и азотная кислота. Известны многие соли для Η4P2O6, так называемые гипофосфаты; напр. для натрия — Na4P2O6∙12H2O, Na3ΗP2O6∙9H2O и Na2H2P2O6∙6H2O, а также известны и двойные соли; изучение солей позволяет считать Η4P2O за сочетание двух фосфорилдигидроксилов — (НО)2ОР—РО(ОН)2, как дитионовая кислота состоит из двух сульфоксилов, а щавелевая из двух карбоксилов [Этилгипофосфат (C2H5)4P2О6 получается при взаимодействии C2Н5J с Ag4Р2О6; представляет густую бесцветную жидкость, которая при перегонке значительно разлагается, а при кипячении с водой разлагается на спирт, фосфорную и фосфористую кислоты.]. Если можно считать доказанным, что двухосновная фосфористая кислота представляет соединение одновалентного остатка ортофосфорной кислоты с водородом, то не будет неожиданностью возможность подобного же сочетания с водородом фосфорилмоногидроксила, двухвалентного остатка той же кислоты — [=PO—OH]; такова, по всей вероятности, одноосновная фосфорноватистая кислота, Н2РО—ОН. Эта кислота получается в форме солей, когда кипятят растворы едких щелочей с обыкновенным Ф., при чем выделяется самовоспламеняющийся на воздухе фосфористый водород; удобнее всего пользоваться едким баритом:
2P4 + 6H2O + 3Ba(OH)2 = 3Ba(O—POH2)2 + 2PH3.
Полученный раствор соли бария фильтруют для отделения от небольшого количества фосфата, осаждают барий надлежащим количеством серной кислоты, сливают прозрачный раствор и осторожно выпаривают, избегая нагревания выше 130°. Полученный таким образом остаток кристаллизуется около 0° и представляет чистую Η3PΟ2, которая плавится при 17,4°. Подобно Η3PO3, фосфорноватистая кислота есть восстановитель и притом более сильный; водород в момент выделения восстановляет ее до РН3, который получается и при нагревании кислоты (выше 140°), именно:
2H3PO2 = HзPO4 + РН3.
Соли ее, или гипофосфиты, МН2PO2, по большей части растворимы в воде, а некоторые и в спирте, и способны кристаллизоваться; они также восстановители, а при нагревании разлагаются, превращаясь в смесь пиро- и метафосфатов с малым количеством красного Ф. и выделяя РН3 и водород. Недавно открытая закись Ф. Р2О могла бы считаться ангидридом фосфорноватистой кислоты, как Р4О6 — ангидрид Η3PΟ3, но ее пока не удалось гидратировать: это желтовато-красный порошок, постоянный при 100°, а при 135° разлагающийся. Что касается недокиси Р4О, то она может быть получена еще следующим образом (о возникновении ее при медленном окислении Ф. упомянуто выше), по новейшим опытам (Michaelis и Pitsch, 1900): 1) если облить очищенный обыкновенный Ф. смесью 1 объема 10 % раствора NaOH и 2 объемов спирта, то он постепенно растворяется при выделении водорода; получается темно-красный раствор, из которого разведенные кислоты и даже СО2 осаждают Р4О; 2) такой же осадок получается из Η3PO2 при ее осторожном обезвоживании, напр. уксусным ангидридом, при чем образуется, кроме того, Н3PO3; превращение выражается следующими уравнениями: 4Η3PO2 — 6Н2O = 2Р2О и Η3PO2 + 2P2O = Н3PO3 + Р4O. Это оранжево-красный или желтый порошок, который в сухом виде может быть довольно сильно нагрет без изменений, но влажный воспламеняется при 90°; при нагревании в индифферентном газе разлагается на Ф. и Р2О5; растворяется в водно-спиртовом растворе NaOH с красным цветом; такой раствор при стоянии или, быстрее, при нагревании выделяет водород и обесцвечивается, при чем получается гипофосфит натрия:
Р4О + 7Н2О = 2Η3PO2 + Н2.
Михаэлис полагает, что все низшие окислы Ф. (ниже Р4О6), описанные в разное время, есть более или менее чистая недокись.
Водородистые соединения, или фосфиды водорода. Кроме газообразного фосфористого водорода РН3, аналогичного с аммиаком, существуют еще жидкий и твердый. Первый содержит водорода в полтора раза меньше и имеет частицу, по аналогии с гидразином и двуйодистым фосфором, вероятно, Η2P—ΡΗ2, или P2H4. Второй еще беднее водородом, а именно содержит его вшестеро меньше, чем РН3; величина частицы его также неизвестна, но ввиду того, что частица фосфора содержит четыре атома, она, быть может, Р4Η2. Свойства РН3 в чистом виде следующие. Это бесцветный газ с запахом чеснока и гнилой рыбы; превращается при —85° в жидкость, которая при —135° замерзает; несколько растворим в воде и сообщает ей особый неприятный, вкус; раствор на свету разлагается при выделении водорода и красного фосфора; такому же разложению подвергается газообразный РН3, если его подвергать действию электрических искр [2], при чем получается полуторный объем водорода. Воспламенить РН3 в воздухе можно только при нагревании (100°), но иногда он загорается и от трения стеклянной пробки при открывании содержащей его склянки. С кислородом РН3 может быть смешан без всякого изменения, но при разрежении такой смеси происходит взрыв — явление, напоминающее о свечении Ф. только в присутствии разреженного кислорода. Горит РН3 весьма ярко, развивая белый дым гидратов Р2О5. Воспламеняется РН3 также от нескольких капель азотной кислоты или хлорной и бромной воды, но в отсутствие воздуха над обыкновенной азотной кислотой, содержащей окислы азота, не воспламеняется; в газообразном хлоре взрывает с сильным звуком и ярким зеленовато-белым светом. По химической функции, как и по составу, РН3 аналогичен аммиаку, но соответствующие аммонийным солям соединения фосфония возникают трудно, а именно для кислородных кислот они совсем неизвестны; хлористый фосфоний РН4Cl получается только при сжимании равнообъемной смеси РН3 и газообразного HCl до 20 атмосфер при 14° (Ogier), бромистый же РН4Br и йодистый PH4J осаждаются при действии РН3 на холодные насыщенные растворы HBr и HJ. Все эти соединения кристалличны; РН4Br при 30° кипит и имеет плотность пара, свидетельствующую о полном разложении на РН3 и HBr; PH4J кипит при 80°, но может быть возгнан и при низшей температуре, при чем получается в виде больших, прозрачных и сильно блестящих кристаллов кубической формы. При простом растворении в воде эти соединения разлагаются, выделяя РН3. Теплота образования из элементов для РН3 4,3 больш. кал., а теплота соединения РН3 и HJ 24,2 б. к. В лабораториях PH4J применяется как могучее восстановительное средство для синтеза содержащих Ф. органических соединений. Жидкий фосфид P2H4 бесцветен, имеет удельный вес 1,01, кипит при 57° — 58° под давлением в 735 мм, не оставляя остатка, если нагревается осторожно. При соприкосновении с воздухом немедленно воспламеняется и ярко горит, подобно свободному Ф.; на свету или при нагревании несколько выше температуры кипения, а также при соприкосновении с малыми количествами соляной или йодистоводородной кислот разлагается по уравнению:
10P2H4 = 12РН3 + 2Р4Н2;
таким образом получается вместе с газообразным и твердый фосфид. Последний представляет желтый порошок без вкуса и запаха; при 170° в токе углекислого газа он разлагается на Ф. и водород, а воспламеняется на воздухе не ранее, как при 160°, так что после промывки водой его можно сушить даже при 140° — 150°. Что касается способов получения, то впервые РН3 был получен (Gengembre, 1783) при кипячении раствора едкого кали с фосфором и оказался самовоспламеняющимся; здесь, кроме того, образуется и гипофосфит калия — по уравнению для едкого барита. Затем РН3 был приготовлен при нагревании фосфористой кислоты (Pelletier, 1790) и не обладал способностью самовоспламенения. Противоречие разъяснилось (Р. Thenard, 1845), когда был открыт жидкий фосфид, ибо присутствие его паров обусловливает самовоспламеняемость [3]. Кроме этой примеси, РН3 может содержать еще и свободный водород, о чем упомянуто по поводу разложения при нагревании солей фосфорноватистой кислоты. К числу способов получения принадлежит разложение водой или кислотами различных металлических фосфидов, каковые обыкновенно аналогичны по составу либо Р2Н4, либо РН3. Обычный фосфористый кальций, который получается при действии паров Ф. на окись кальция и представляет темно-бурые куски, состоящие из фосфида и пирофосфата кальция (14CaO + 14P = 5Р2Ca2 + 2Ca2P2O7), дает при действии теплой (60°) воды в атмосфере водорода жидкий фосфид: Р2Са2 + 4H2O = P2H4 + 2Ca(OH)2, который уносится в виде пара водородом и сжижается в охлажденном (0°) приемнике; при этом разложение на РН3 и Р4Н2 сравнительно незначительно; но, употребляя для разложения соляную кислоту, можно получить главным образом РН3 с примесью Р2Н4 и вследствие того самовоспламеняющийся. Такой РН3 можно лишить этой способности, очистив от Р2Н4 путем пропускания чрез сильно охлажденную U-образную трубку, где Р2Н4 и остается. Недавно полученный (Moissan, 1899) кристаллический, красно-бурого цвета фосфористый кальций Р2Ca3, для чего нужно накалить смесь 310 весов. частей Са3(РО4)2 с 96 в. ч. угля в электрической печи током в 950 ампер и 45 вольт в течение 4 минут, дает с водой HP3, не воспламеняющийся произвольно. Лучший путь приготовления чистого РН3 — это разложение раствором едкого кали йодистого фосфония:
РН4J + КОН = РН3 + H2O + KJ.
Равномерный ток РН3 получают также при действии обыкновенного серного эфира (содержащего воду) на РН4J, причем HJ удерживается в виде HJ∙2(С2H5)2О; операцию можно вести в маленьком аппарате Киппа. Самый РН4J готовится так: 400 гр. обыкновенного Ф. растворяют в таком же количестве сероуглерода; к раствору при хорошем охлаждении постепенно прибавляют 680 гр. йода; затем отгоняют растворитель и остаток, пропуская струю углекислого газа, — операция ведется в реторте, соединенной с широким холодильником, — постепенно смешивают с 250 гр. воды; реакция:
5J + 9P + 16H2O = 5РН4J + 4Н3РО4
идет с выделением тепла, так что кристаллический возгон PH4J осаждается на стенках холодильника; в конце необходимо подогревать; часть Ф. превращается в красный Ф., почему и берут избыток. Подобно аммиаку, РН3 способен соединяться с некоторыми хлористыми металлами, например 3AlCl3∙РН3, 3SnCl4∙2РН3 — соединения, разлагаемые водой; раствор CuCl в соляной кислоте очень быстро поглощает РН3, причем сначала осаждается кристаллическое соединение CuCl∙PH3, затем жидкое CuCl∙2PH3. При взаимодействии с фосгеном, растворенным лучше всего в бензоле или хлороформе, получается аналог мочевины:
COCl2 + 2РН3 = СО(РН2)2 + 2HCl,
желтый порошок, нерастворимый в обычных растворителях и назначаемый для медицинских целей (1896, патент фабрики Meister, Lucius and Brüning in Höchst am M.). При действии РН3 на растворы металлического К или Na (см. Калий, Натрий) в жидком аммиаке происходит (Joannis, 1894) при выделении водорода образование гидрофосфидов этих металлов; испаряя аммиачные растворы, получают белые иголочки КРН2 или бесцветный твердый NaPH2, который всегда содержит Na3Р. При нагревании гидрофосфиды превращаются в фосфиды:
3КРН2 = K3P + 2РН3;
водой все эти соединения разлагаются на едкую щелочь и РН3. При взаимодействии аммиачных растворов гидрофосфидов с закисью азота она превращается в равный объем азота. Если подействовать на металлический натрий РН3 при нагревании, а затем, не охлаждая трубки, чистой окисью углерода, то получается самовоспламеняющийся твердый продукт (Schober и Spanutius, 1894), который с безводным спиртом дает темно-красный раствор, а водой разлагается так, как будто бы это был цианистый натрий, в котором Ф. заместил азот:
NaCP + 2Н2О = HCl2Na + РН3,
получается муравьинокислый натрий и РН3 — вместо NH3, как это имеет место для цианистого натрия, который может быть приготовлен действием окиси углерода на NaNH2. Эта способность РН3 давать с натрием и калием гидрофосфиды и даже фосфиды сближает его еще более с аммиаком, а кроме того, содействует утверждению вышеизложенного представления о строении фосфористой кислоты, как содержащей один водород непосредственно при Ф.
Сульфиды Ф. Прежде полагали, что сера и Ф., как элементы сходные, не образуют определенных соединений; но затем оказалось, что сера способна играть роль кислорода по отношению к Ф. и дает с ним соединения, образующиеся с большим выделением тепла и аналогичные по составу и отношениям с окислами (Берцелиус). Сера и Ф. образуют растворы (до 25 % серы), если сплавлять их под водой или нефтью, не нагревая выше 70°; такие растворы остаются иногда жидкими и при обыкновенной температуре; Ф. из них может даже кристаллизоваться. Но при более сильном нагревании происходит весьма экзотермическое взаимодействие, так что необходимо умерять реакцию, пользуясь красным Ф. вместо обыкновенного и серой в виде кусочков, а не в порошке; оперируют при малом доступе воздуха или в токе углекислого газа с предварительно заготовленными смесями в той или иной пропорции, смотря по соединению, к которому стремятся. Известны следующие соединения: P4S3 кристаллизуется из сероуглеродного раствора в виде желтых призм, плавится при 166°, кипит при 380°; P4S6, трисульфид, кипит при 490° и мало растворим в сероуглероде; P3S6, дисульфид, плавится при 296—298° и способен улетучиваться; Р2S5, пентасульфид, кристаллическая желтоватая масса, плавится при 274° — 276° и кипит при 518°. Все эти соединения водой разлагаются, при чем образуется сероводород, фосфорная или фосфористая кислота, а также и PHЗ — при малом содержании серы:
P4S3 + 9Н2O = 3Н2S + 3Н3PO3 + РН3,
Р2S5 + 8H2O = 5Н2S + 2Н3PΟ4;
особенно охотно так реагирует Р2S5, напоминая о PCl5. В лабораториях находит применение P4S3, при приготовлении тиофена, и P2S5, наприм. для получения из спирта меркаптана. Тиоангидридные свойства принадлежат сульфидам Ф. При взаимодействии Р2S5 с PCl5 получается хлористый тиофосфорил SPCl3, бесцветная жидкость, которая дымит на воздухе, кипит при 125° и имеет при 298° плотность пара, отвечающую формуле; с едким натром из нее получается тиофосфат Na3PSO3.
Азотсодержащие соединения. Непосредственно с азотом Ф., как упомянуто, не соединяется; соединений, содержащих только эти элементы, вообще неизвестно; но соединения, в состав которых входят еще и другие элементы, многочисленны — не считая аммонийных солей описанных кислот Ф. — и хотя изучаются уже давно (Gladstone, Schiff, Gerhardt и друг.), все еще не вполне расследованы. По новейшим данным (Н. N. Stokes, 1893—1899) амидоортофосфорная кислота, или фосфаминовая Н2N∙PO(ОН2), получается так: нагревают, с обратнопоставленным холодильником, смесь OPCl3 с фенолом, до полного удаления HCl; остаток Cl—PO(O—C6H5)2 разбавляют абсолютным спиртом и прибавляют до щелочной реакции спиртового раствора аммиака —
NH3 + Cl—PO(O—C6H5)2 = H2N—PO(O—C5H5)2 + NH4Cl;
затем извлекают нашатырь водой и очищают фенольный эфир, многократно кристаллизуя из спирта или хлороформа; при омылении этого эфира едким кали получается калиевая соль фосфаминовой кислоты; свободная же кислота была выделена сероводородом из свинцовой соли. Н2N—PO(ОН)2 кристаллична, нерастворима в спирте, легко растворима в воде и имеет сладкий вкус и кислую реакцию; дает одно- и двуметаллические соли, из коих первые, для металлов щелочей, имеют среднюю реакцию и получаются из растворов вторых при действии уксусной и даже угольной кислот. Однометаллические соли при прокаливании дают метафосфаты, двуметаллические — пирофосфаты; но Н2N—PO(OAg)2 при 180° превращается в соль, вероятно, пиримидокислоты — HN=[PO(OAg)2]2, которая по сплавлении представляет желтое, растворимое в нашатырном спирте, стекло. Диамидоортофосфорная кислота (Η2N)2PΟ—ΟΗ получена подобным же путем из Cl2PO—O—C6H5; она кристаллична; в сухом виде, подобно предыдущей кислоте, постоянна; но растворенная легко, как и та, заменяет свои амиды гидроксилами, в особенности в присутствии иной кислоты; в щелочных же растворах гидролиз идет очень медленно даже при кипячении. Азотистая кислота превращает ее на холоде в амидокислоту, а затем в ортофосфорную:
HNO2 + (Н2N) = Н2O + N2 + (НО.).
Серебряная соль (H2N)2PO—OAg хорошо кристаллизуется; но, кроме того, получены еще соли с 2, 3, 4 и 5 атомами серебра, которые содержат, конечно, серебро вместо водорода амидогрупп, а некоторые могут считаться происшедшими из особой пятиосновной кислоты (Н2N)2P(OH)3. Эта кислота могла бы получиться из (Н2N)2PO—OH через присоединение воды, но неизвестна в свободном виде; желтая серебряная соль имеет такой состав и, быть может, строение (H2N)2P(OAg)3, а похожая на свежеосажденную окись железа — (AgHN)2P(OAg)3. Последняя соль взрывает уже при растирании в ступке; другие же серебряные соли взрывают более или менее сильно только при нагревании. Триамид (H2N)РО (Schiff, 1857), который должен получаться путем полного насыщения Cl3РО аммиаком и промывки полученной смеси с NH4Cl водой, позднейшим исследователям (Gladstone, Mente) получить не удалось. Имидоамид или фосфамид Жерара HN=PO—NH2 — продукт действия NH3 на PCl5, затем воды и КОН или К2СО3, оказался (Gladstone) неоднородным; при нагревании он превращается в нитрил NPO, белый аморфный порошок, который плавится при красном калении и застывает затем в черную стекловатую массу, а при сплавлении с КОН превращается в Κ3ΡΟ4 и NH3; подобное превращение для имидоамида имеет место уже при кипячении с раствором КОН. Изучая отношение Cl3РО, а также PCl5 и Р2O5 к NH3 и затем к воде, пришли (Gladstone, 1864—1869) к целому ряду кислот, которые имеют строение неполных амидов пирокислот — обыкновенной и произведенной из четырех частиц ортокислоты Н4Р2О7 и Н6Р4О13. Но наиболее изученные из этих имидокислот представляют (Mente, 1888), быть может, диметафосфорную кислоту, в коей один или два кислорода замещены через (=NH), т. е. это имидокислоты, именно HN=(P2O3)(OH)2 и (HN=)2(P2O2)(OH)2 с отвечающей амидокисл. (HN=)2(P2O2)(NH2)(OH). Хлорангидрид первой имидокислоты, но не HN=(P2O3)Cl2, a HN=(POCl2)2, при 290° превращается в![]()
по уравнению: 3HN(POCl2)2 = 2N(POCl2)3 + NHЗ, откуда, действуя водой, можно перейти к новой кислоте:

Это нитрилометафосфорная кислота, для которой получены многие соли; сама кислота очень растворима в воде, как и соли ее с металлами щелочей [При своих опытах с Cl3PO Менте пользовался вместо NH3 преимущественно карбаматом аммония, NH4—O—CO—NH2.]. В близком отношении к этим кислотам находятся недавно открытые (Stokes) метафосфиминовые кислоты — (HN=PO—OH)n, a также и (HN=РО—ОН)n∙Н2O. Хлористый фосфонитрил (NPCl2)3 (Либих) стоит в прямой связи с этими кислотами; он получается вместе с другими соединениями того же состава, но различной степени полимеризации (Stokes), лучше всего путем нагревания смеси PCl5 и нашатыря (частица на частицу: PCl5 + NH4Cl = NPCl2 + HCl) в запаянной трубке и перегонки нерастворимой в газолине части продукта. В настоящее время известны (NPCl2)n, где n = 3, 4, 5, 6 и 7; все они при 250° — 350° полимеризуются в камедеобразный полимер (NPCl2)x, но при более сильном нагревании происходит обратное превращение. Давно известный простейший полимер (NPCl2)3 кристаллизуется в прозрачных табличках, похож на камфару, плавится при 114°, кипит при 250°; в воде он нерастворим и хорошо летуч с ее парами; растворим в спирте, хлороформе и особенно в эфире. Почти таков же и другой полимер (NPCl2)4, но плавится он при 123,5°, кипит при 328,5° и очень мало летуч с парами воды. Ни кипящая вода, ни кислоты, ни щелочные растворы не действуют на эти соединения; но продолжительное и непрерывное взбалтывание эфирных растворов с водой приводит, однако, к замещению хлора элементами воды, сначала отчасти (хлоргидрины), а затем и вполне:
NPCl2 + 2H2O = NP(OH)2 + 2HCl
и, вероятно,
NP(OH)2 = ΗΝ=PO—OH.
Получаются таким образом из каждого полимера упомянутые кислоты с строением кольцеобразнозамкнутым (ΗΝ=PO—OH)n или открытые (ΗΝ=РО—ОН)n∙Н2О; первые аналогичны, вероятно, полиметафосфорным кислотам и отличаются от них тем, что содержат на каждый атом Ф. одну имидогруппу вместо кислорода. Триметафосфиминовая кислота (ΗΝΡΟ2Η)3 из обычного хлористого фосфонитрила шестиосновна и дает соли трех-, четырех- и шестиметаллические. Кислота (HNPO2H)4 наиболее устойчива и дает соли двух-, четырех- и восьмиметаллические. Если осторожно нагревать PCl5∙8NHЗ, продукт присоединения аммиака к PCl5 в CCl4 растворе, то удаляется много аммиака, затем гонится NPCl2 — вероятно, один из полимеров хлористого фосфонитрила, с темп. пл. 106° и возгонки 175° — 200°; при более сильном нагревании отгоняется нашатырь и остается так назыв. фосфам ΡN2H (Besson). To же самое соединение, известное уже давно (Liebig и Wöhler), получается и в различных других условиях из PCl5, P2S5 и пр.; вероятно, оно возникает так:
PCl5 + 6NH3 = N=PCl—NH2 + 4NH4Cl
и N=PCl—NH2 = N=P=NH + HCl.
Частица фосфама, по всей вероятности, очень полимерна (PN2H)X; это белый порошок, который нерастворим в воде и в других растворителях, не плавится и не улетучивается даже при красном калении, но при нагревании в присутствии воздуха окисляется, выделяя белый дым. При сплавлении с влажным едким кали фосфам разлагается так:
ΡΝ2H + 3ΚΟΗ + Н2O = Κ3PΟ4 + 2ΝΗ3,
при чем выделяется свет и тепло; нагревание же с водой приводит к метафосфорной кислоте и аммиаку. Фосфам — полимерный имидонитрил ортофосфорной кислоты, так как с водой при сравнительно низкой температуре он дает метакислоту, ортокислоту же — только при избытке щелочи и при более высокой температуре. Если действовать на фосфористый ангидрид в эфирном или бензольном растворе газообразным аммиаком, то получается (Thorpe и Tutton, 1891), вероятно, диамид фосфористой кислоты в смеси с фосфидом аммония, в виде белого порошка:
P4O6 + 8NH3 = 3HPO(NH2)2 + HPO(NH4)2.
Этот порошок растворяется в воде с таким выделением тепла, что раскаляется; с довольно крепкой соляной кислотой реагирует весьма энергично, выделяя несамопроизвольно воспламеняющийся фосфористый водород и свободный Ф., в растворе же получается нашатырь, фосфористая и фосфорная кислоты. О соединении Ф. с металлами и о фосфорнокислых солях — см. соответствующие металлы.
Фосфор (аллотропия; см. Изомерия). — Ф. полиморфный элемент; он известен в нескольких видоизменениях, из которых полнее исследованы два: Ф. белый и красный; менее изучены Ф. оранжевый (может быть, их несколько) и Ф. металлический (может быть, не что иное, как крупнокристаллический красный Ф.). Белый Ф. (он же желтый, бесцветный, обыкновенный Ф. — различных учебников) получается при быстром охлаждении перегретых паров Ф., а потому представляет главную массу продажного Ф. Бесцветное, вполне прозрачное кристаллическое (правильные октаэдры с сильным стеклянным блеском [Микроскопические правильные додекаэдры из бензола (Ретгерс).]) вещество, постепенно желтеющее и тускнеющее на свету; при продолжительном действии света он сильно краснеет, а иногда даже и чернеет [Обыкновенно в Ф., даже чистом на вид, содержатся некоторое количество серы и мышьяка. Различное отношение Ф. к свету может быть обусловлено примесями (см. ниже).], превращаясь отчасти в красный. На холоде белый Ф. хрупок, но выше +15° Ц. он мягок, легко чертится ногтем (твердость 0,5 шкалы Моса [Ридберг]), и палочка белого Ф. может быть, не ломаясь, изогнута несколько раз; примеси делают его хрупким. Уд. вес белого Ф. при 10° — 1,83; темп. пл. 43,94° Ц. (при давлении 1 атм; Тамман); темп. кип. 290° Ц. (Троост и Готфейль), как видно из таблицы давлений пара жидкого белого Ф., где t — град. Ц. и Р — давление в см.
| t° | 165° | 180° | 200° | 219° | 230° | 290° |
|---|---|---|---|---|---|---|
| P | 12 | 20,4 | 26,6 | 35,9 | 51,4 | 76,0 |
| t° | 360° | 440° | 494° | 503° | 511° | |
| P | 3,2 атм. | 7,5 | 18,0 | 21,9 | 26,2 |
Эти числа отвечают моновариантной системе: Ф. белый, жидкий + пар. Темп. пл. с увеличением давления повышается (объем твердого белого Ф. меньше объема жидкого белого Ф.); по Тамману, след. образом:
| Темп. пл. | 43,94° | 97,4° | 100,18° (438°) |
|---|---|---|---|
| Давление в атм. | 1 | 2000 | 2155 (14400) |
Числа этих рядов дают: первые — абсциссы, а вторые — ординаты пограничной кривой моновариантной системы из двух сосуществующих фаз: белый Ф. жидкий + белый Ф. твердый. Числа: 438° и 14400 атм. экстраполированы Тамманом на основании предыдущих; они представляют максимальную возможную температуру плавления белого Ф. при условии равенства объемов твердой и жидкой фаз. Уд. вес пара белого Ф. при температуре кипения = 4,42 (по воздуху) и остается таковым почти до 900°; затем он постепенно падает и при 1400° Ц. = 3,147. Белый Ф. почти не растворим в воде [Некоторой растворимостью он все же, кажется, обладает, а именно = 0,1 гр. в 500 к. с. (Бокорный); вода, взболтанная с Ф., вызывает явления отравления, но таким свойством, может быть, обладают низшие степени окисления Ф., избежать присутствия которых трудно, или же Ф. растворяется в виде гидрата, способного существовать, по Ретгерсу.] и трудно — в эфире, спирте и скипидаре; очень легко растворим в сероуглероде и треххлористом Ф.; кроме того, Ф. растворим в йодистом метилене (Ретгерс), трехбромистом Ф. (Шенк), жидком аммиаке (Гюго) и жидком сернистом ангидриде (Вальден). Красный Ф. открыт Шрёттером (1845), наблюдавшим образование его при долговременном нагревании при 280° белого Ф.; он же наблюдал, что при более высокой температуре красный Ф. может обратно превращаться в белый Ф. Явление исследовалось затем Гитторфом, Лемуаном, Троостом и Готфейлем. Первый (1865) нашел, между прочим, что превращение идет быстрее при температурах, лежащих выше 300° (следовательно, выше темп. кип. белого Ф. под давлением большим, чем 1 атм.) и что оно связано с выделением тепла; благодаря последнему обстоятельству превращающаяся при 300° масса Ф. может самопроизвольно нагреться до 400° [Цифровые данные Гитторфа не могут претендовать на большую точность.]. Второй (1871) установил, что если взять уже готовый красный Ф. и нагревать его в замкнутом сосуде при какой-нибудь определенной температуре, напр. при 440° (темп. кип. серы), то уд. вес образующаяся пара растет по мере увеличения массы нагреваемого Ф.; при упомянутой температуре уд. вес перестает увеличиваться с прибавлением Ф., достигает maximum’a, когда в 1 литре заключается 4,59 гр. пара; но если нагревание продолжается долговременно, то наблюдается новое падение количества пара, останавливающееся при содержании 3,72 гр. в 1 литре (через 47 час., Троост и Готфейл). Если же исходить из белого Ф., то первоначальные давления его пара несравненно выше, а затем по мере образования красного Ф. эти давления падают, пока не достигнут постоянной величины, тождественной с давлением при тех же условиях красного Ф. Троост и Готфейл (1873) измерили давления последнего и нашли следующие величины:
| t° | 360° | 440° | 487° | 510° | 531° | 550° | 570° |
|---|---|---|---|---|---|---|---|
| Р (в атм.) | 0,12 | 1,75 | 6,8 | 10,8 | 16,0 | 31,0 | 56,0 |
Вместе с Лемуаном они на них смотрят как на данные, определяющие условия превращения красного Ф. в белый, но, согласно Б. Роозебоому (ср. В. Roozeboom, «Heterogene Gleichgewichte vom Standpunkte der Phasenlehre», кроме того, Банкрофт, «The Phase Rule», и Дюгем, «Thermodynamique et Chimie»), гораздо проще и вернее принимать, что эти цифры отвечают моновариантной системе из твердого красного Ф. и пара Ф. Аномалия, наблюдавшаяся Лемуаном и заключавшаяся в постепенном падении давления пара Ф. (см. выше), по Троосту и Готфейлю, объясняется тем, что существуют на самом деле несколько видоизменений красного (?) Ф.; а именно получаемый при 265° он представляет массу ярко-красного цвета, напоминающую реальгар (As2S5); Ф., получаемый при 440°, имеет оранжевый оттенок, а при 500° — обладает ясным фиолетовым оттенком. Эти видоизменения Ф. отличаются, кроме того, по Троосту и Готфейлю, различными удельными весами и теплотами горения. Нельзя, однако, их считать окончательно установленными (Ретгерс, Шенк) и возможно, что они представляют смеси в разных отношениях красного Ф. с металлическим (см. ниже). Красный Ф. может быть получен не только при нагревании белого Ф. или под каталитическим влиянием света, но и под влиянием некоторых химических каталитических агентов; так, напр., ничтожные количества йода или селена, действуя на сероуглеродный раствор Ф., вызывают быстрое выпадение из него красного Ф. Если йод брошен в массу расплавленного Ф., нагретого до 180° (в атмосфере CO2), то превращение белого Ф. в красный происходит почти мгновенно. Красный Ф. (в очень многих учебниках он же аморфный Ф.) представляет кристаллическую массу (Ретгерс, Линк) с уд. весом, колеблющимся между 2,15 и 2,34; он плавится, по Чапману, около 630° при неизвестном пока давлении. Красный Ф. почти совершенно нерастворим в сероуглероде и в других растворителях белого Ф.; в трехбромистом Ф. (в 100 в. ч.) при 172° его растворяется всего 0,2601 в. ч., а при 184° — 0,3634 в. ч. (Шенк); едва заметно растворим красный Ф. и в водном едком кали (Бёрджес и Чапман); химическая энергия его сравнительно с таковой же для белого Ф. — ничтожна; он не ядовит. Следующая схематическая диаграмма (фиг. 1), принадлежащая Б. Роозебоому, в высшей степени наглядно передает вышеизложенное.

На ней линия CO2 отвечает моновариантной системе: Ф. белый твердый + пар, линия O2E — то же моновариантной системе, Ф. белый твердый + Ф. белый жидкий) и кривая О2Е представляет отрезок изученной Тамманом кривой, выражающей изменения температуры плавления твердого белого Ф. с давлением; линия О2О1А есть пограничная кривая моновариантной системы: белый Ф. жидкий + пар; точка О2 отвечает нонвариантной системе: белый Ф. твердый + жидкий + пар, т. е. она представляет темп. плавления белого Ф. под давлением его собственного пара. Кривая ВО есть пограничная кривая моновариантной системы: красный Ф. (предполагается, что возможно только одно видоизменение его) + пар; точка Ο1 отвечает температуре плавления красного Ф. и кривая OF представляет еще совершенно не изученную пограничную кривую моновариантной системы: красный Ф. твердый + жидкий (белый?), или же изменение температуры плавления красного Ф. с нарастанием давления; наконец, кривая Ο1А представляет кривую моновариантной системы: Ф. (белый?) жидкий + пар и вместе с тем продолжение кривой O2O1. На диаграмме ясно видно, что давление пара белого Ф. при низких температурах значительно превышает давление пара красного Ф. и что по мере возрастания температуры разность их уменьшается. Кроме того, диаграмма, как видно, предполагает, что белый Ф. находится в том же отношении к красному Ф., в каком, например, находится переохлажденная жидкая вода к воде твердой (льду, см. Правило фаз); это предположение объясняет ряд фактов, наблюдаемых при взаимных переходах белого и красного Ф. Так, напр., благодаря исследованиям Таммана известно, что превращение переохлажденных жидких систем в твердые идет в общих чертах следующим образом: скорость превращения, минимальная при инверсионной точке (см. Правило фаз), т. е. при температуре плавления твердой системы, начиная с некоторой степени переохлаждения, быстро возрастает, затем переходит через максимум, наблюдаемый в более или менее широких пределах температуры, а потом быстро падает. Почти все это наблюдено для превращения белого Ф. в красный; здесь при обыкновенных условиях нельзя уловить превращения, потому что благодаря громадному переохлаждению скорость его близка к нулевой (ср. расчет Оствальда в его «Grundlinien der anorganischen Chemie», 1900, 359); с повышением температуры она растет (Гитторф, Лемуан) и выше 511° уже становится настолько значительной (близкой к maximum’y?), что тут совершенно невозможно наблюдать давление пара белого Ф., превращающегося почти мгновенно в красный; неизвестно, следовательно, только падение скорости превращения при приближении к точке О1. Затем — превращение переохлажденной жидкости в твердое тело представляет экзотермический процесс: выделение тепла констатировано и для превращения белого в красный Ф. (см. выше); Мутье (1874) нашел даже, что применяя к обеим кривым 02А и ВО уравнение Клапейрона-Клаузиуса — Т(dp/dt) = Q/dr [Т — абсолютная температура опыта, dp/dt нарастание давления пара с температурой, Q — тепло превращения, отнесенное к единице веса превращающегося тела, a dr — изменение объема системы при превращении (разность объемов насыщенного пара и твердого, жидкого тела).], можно вычислить теплоту, выделяющуюся при превращении белого Ф. в красный и довольно хорошо согласующуюся с калориметрическими данными. Диаграмма же позволяет легко проследить и за условиями превращения одного видоизменения Ф. в другое. Выше указано, что с увеличением количества нагреваемого красного Ф. (при данном объеме прибора) давление его пара быстро падает; процесс отвечает пунктирной прямой GH, и падения давления должно ожидать на основании закона Ле-Шателье (см. Правило фаз), гласящего в этом частном случае, что увеличение концентрации перемещает равновесие в сторону системы, занимающей меньший объем и обладающей, следовательно, меньшим давлением. Затем можно легко представить, что при быстром охлаждении пара мы имеем дело с процессом, отвечающим пунктирной линии HJ [Нет необходимости, чтобы она была параллельна линии абсцисс, как это представлено на диаграмме, и ордината точки J может быть немного меньше ординаты точки Н], которая показывает, что пар точки H перегрет по отношению к пару точки J, и объясняет возможность конденсации в J белого Ф. Такое представление на первый взгляд может казаться противоречащим данным Лемуана относительно скорости превращения красного Ф. в белый, каковая, по его убеждению, ничтожна; надо, однако, заметить, что числа Лемуана представляют не действительно измеренные величины, а скорости, вычисленные в предположении, что наблюдавшиеся им равновесные состояния являлись следствием двух процессов с противоположными знаками; выше же разъяснено, что его измерения относятся просто к равновесию красного Ф. с паром, но не к превращению красного Ф. в белый; а во-вторых, тот же Лемуан наблюдал гораздо большие скорости, когда он нагревал красный Ф. в присутствии металлической меди, способной в условиях опыта соединяться с белым Ф., если бы он образовывался. След., его данные не противоречат развиваемому взгляду, а с другой стороны он подтверждается прямыми опытами Трооста и Готфейля, показавшими, что при том давлении, при котором белый Ф. оставался в виде капель в конце трубки, нагретом до 324° Ц., красный Ф. осаждался в виде твердого тела в противоположном конце той же трубки, нагретом до 350° Ц.; очевидно, что опыт мыслим и в обратном порядке, а это как раз то, что выражает линия HJ [Наблюденная тут Троостом и Готфейлем перегонка вещества из пространства с низшей температурой в пространство с более высокой температурой характерна для превращения переохлажденной жидкости в твердое тело (см. В. Roozeboom, "Heterogene Gleichgewichte etc. ", стр. 68, прим. 2, и Duhem, «Thermodynamique et Chimie», стр. 204). Очевидно, что при некотором подборе температур тут мыслимо и равновесное состояние, т. е. отсутствие превращения.]. Что перечисленными исследованиями еще далеко не закончено изучение Ф., ясно видно на проекции диаграммы Роозебоома на плоскость температуры и объемов. Линия, идущая из точки B, представляет схематически кривую расширения красного Ф. с повышением температуры; она не изучена; ее точка О соединена вертикальной прямой с кривой O2А, чтобы показать, что красный Ф. плавится с расширением, что пока вероятно; кривая СО2 отвечает кривой расширения твердого белого Ф., а кривая О2О1А представляет схематическую кривую расширения жидкого белого Ф., тоже еще не изученную и которую экспериментально, несомненно, трудно установить; точка А должна бы представлять неизвестную критическую температуру Ф.; в ней кривая объемов жидкого Ф. должна встретиться с кривой объемов насыщенных паров Ф., но последняя пока тоже не исследована, и даже неизвестно, должна ли она быть в единственном числе, как она нанесена на диаграмме, или же красному Ф. должна отвечать своя собственная кривая объемов насыщенного пара; судя по Арктовскому, который наблюдал при низких температурах (ниже 266°) и давлениях возгонку красного Ф., последнее возможно; оно даже необходимо, если закон прямолинейного диаметра Кальете-Матьаса (см. Газы) общеприменим. Таким образом, отношения, наблюдаемые между красным и белым Ф., являются совершенно отличными от того, что известно для отношений серы октаэдрической (ромбоэдрической) и призматической (одноклиномерной); ср. Правило фаз. Белый Ф. малоустойчив по отношению к красному Ф., не только в точке О1 (см. диаграмму), но и при целом ряде температур, а между тем опыт показывает, что при обыкновенной температуре непосредственное соприкосновение белого Ф. с красным еще не вызывает сколько-нибудь заметного превращения первого, и во-вторых, нет температуры, при которой одно твердое видоизменение нацело переходило бы в другое видоизменение, не переходя предварительно через парообразное состояние. На эти различия уже давно обратил внимание Леман, предложивший называть энантиотропными изомерные, вернее, аллотропные состояния, подобные состояниям упомянутых видоизменений серы, и монотропными — тела, подобные обоим видоизмнениям Ф. Следующая диаграмма уясняет различие этих систем (Шенк и Шнейдер, Оствальд). Пусть кривые АА и ВВ (фиг. 2) представляют кривые давлений насыщенных паров двух твердых изомерных (аллотропных) видоизменений данного тела; благодаря различию их угловых коэффициентов они пересекаются в точке Р.

Здесь давление паров обоих видоизменений тождественно, и между ними возможно и должно иметь место равновесие; P есть, следов., точка превращения (инверсионная точка); выше ее видоизменение А, как малоустойчивое, переходит в В; ниже — наблюдается обратное явление. Кривая давления насыщенных паров жидкого тела может лежать или выше точки P (СI), или ниже ее (СII), или, наконец, совпадать с ней (СIII). Последний случай пока неизвестен. В первом — при нагревании видоизменения А мы наблюдаем в точке P переход его в видоизменение В и при дальнейшем нагревании достигаем точки S, представляющей температуру плавления видоизменения В; при охлаждении наблюдаются те же явления в обратном порядке; если удается избежать превращения в точке Р, то выше ее мы имеем дело с перегретым видоизменением А, плавящимся при температуре S″; S′ лежит ниже S, т. е. малоустойчивое при высокой температуре видоизменение, но устойчивое при обыкновенной, плавится ниже устойчивого при высокой температуре. Все это целиком приложимо к ромбоэдрическому и одноклиномерному видоизменениям серы и характеризует энантиотропные системы. Если же кривая давления насыщенного пара жидкости пересекает кривые АА и ВВ ниже точки РI, то, нагревая тело А, мы приходим к его температуре плавления S′, а, нагревая тело В, мы приходим к его температуре плавления S′; оба видоизменения плавятся ниже точки перехода, и потому в твердом состоянии они не могут быть превращены одно в другое простым соприкосновением; с кривой давления жидкости они находятся в равновесии только при соответственных температурах плавления, а потому взаимное превращение при температурах, близких к S, возможно только при конденсации более холодного пара В на более нагретом теле А, т. е. должно идти очень медленно и неправильно; при всех температурах видоизменение В переохлаждено по отношению к видоизменению А, температура плавления S менее устойчивого видоизменения тут лежит ниже темп. плавления S′ более устойчивого видоизменения. Все это наблюдается для белого и красного Ф. и характерно для монотропных систем [Более сложным примером монотропных систем являются, напр. насыщенные растворы различных гидратов сернокислых солей (ср. Правило фаз о растворимости Na2SO4∙7H2O и Na2SO4∙10Н2О); роль кривой давления пара жидкой фазы играет тут кривая растворимости безводной соли, с которой одной эти гидраты и находятся в равновесии в точках пересчений кривых растворимости. И тут менее устойчивая система (гидрат) начинает выделять безводную соль при более низкой температуре, чем система (гидрат) более устойчивая; далее, чем менее устойчива система, тем она более растворима, и т. д. См. еще Фумаровая кислота.]. Металлический Ф. Под этим названием описано Гитторфом видоизменение Ф., полученное им при растворении под давлением при 600° (в отсутствие воздуха) Ф. в расплавленном свинце, каковой по охлаждении раствора был удален слабой азотной кислотой. По Гитторфу, металлический Ф. обладает серым металлическим цветом, кристалличен и еще более химически индифферентен, чем красный Ф. Этих данных недостаточно, чтобы признать металлический Ф. за особое аллотропическое (тоже монотропное?) видоизменение (ср. наблюдения Ретгерса), но нельзя не обратить внимания на то, что металлическое видоизменение Ф. вероятно по близкому сходству Ф. с мышьяком и существованию металлического мышьяка и по положению Ф. в периодической системе элементов (см. Периодический закон) в соседстве с Углеродом и Кремнием, которые оба известны не только в металлоидных (алмазовидных), но и в металлических (графитовидных) видоизменениях.
- ↑ Правда, многие соли тяжелых металлов, если прибавить к их растворам фосфористой кислоты, не осаждаются едким натром, образуя двойные растворимые соли, напр. для медного купороса — NaCuPO3 (Vanino, 1899), но это, конечно, особые двойные соли, подобные тем, которые получаются при растворении окиси меди в щелочных растворах амидокислот или оксикислот. Как при действии уксусного ангидрида (СН3—СО)2О или хлорангидрида CH3—COCl из амидокислот получаются кислоты, содержащие ацетил на месте водорода при азоте, так и для фосфористой кислоты возможно ацетильное производное. — H. А. Меншуткин (1866), действуя хлористым ацетилом, получил из нее двухосновную ацетилпирофосфористую кислоту —
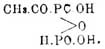
- ↑ Нужно только, чтоб разряд происходил между угольными электродами, так как платиновые быстро сплавляются, превращаясь в серебристый фосфид.
- ↑ Фосфористые водороды, подобно H2S, NH3, СН4, образуются при гниении растительных и животных отбросов (гнилой сыр иногда пахнет чесноком), этим, быть может, объясняются болотные огни.